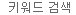Low cell number ChIP-seq using the ThruPLEX DNA-Seq Kit as a tool for epigenetic profiling
Data kindly provided by: Yanina Bogliotti and Pablo Ross at University of California, Davis, California, United States
◆ Introduction
Polymerase, transcription factor, chromatin modifier와 같이 DNA와 결합하는 단백질은 DNA의 복제부터 유전자의 발현이나 염색질 구조를 조절하기 위해 DNA를 수정하는 과정까지 세포의 다양한 필수 세포 기전에 중요하게 작용한다. DNA와 결합하는 단백질은 복제 개시점이나 프로모터와 같은 특정 genomic loci에 결합하거나, single stranded DNA에 결합하는 단백질, helicase, polymerase와 같이 다양한 서열에 따라 DNA에 결합할 수도 있다 (Farnham 2009; Johnson
et al. 2007 ; Martin과 Zhang 2005). 많은 연구자들은 histone과 nucleosome의 위치와 변형이 유전자 조절에 어떤 역할을 수행하는지에 대한 관심이 매우 높다 (Barski
et al. 2007). 대표적으로, Lysine 4에 위치한 histone 3의 tri-methylation은 결합하고 있는 loci의 전사를 촉진하는 것으로 알려진 반면, lysine 27에 위치한 histone 3의 tri-methylation은 전사를 감소시키는 것으로 확인되었다 (Martin and Zhang 2005). 크로마틴면역침강법 (Chromatin immuno precipitation; ChIP)은 DNA와 결합하는 단백질과 이들이 타겟으로 하는 게놈간의 상호 작용을 연구하기 위해 사용되는 아주 효과적인 방식이다. NGS 기술이 점차 발전함에 따라, 과학자들은 high-throughput DNA-seq과 ChIP-seq을 결합하여, 게놈 전체에서 DNA와 결합하는 단백질의 표적 염기 서열을 보다 쉽게 확인할 수 있게 되었다.
ChIP-seq 실험의 일반적인 과정은 아래와 같다 (
그림 1).
1) Formaldehyde와 같은 reversible cross-linker를 처리해 단백질이 DNA에 결합하도록 한다.
2) Sonication이나 효소 처리를 통해 DNA를 절단한다.
3) Magnetic beads에 부착되어 있는 특이 항체를 이용해 protein-DNA complex를 침전 시킨다.
4) 단백질로부터 결합된 DNA를 떼어내고, 이 DNA로 library를 제작한다.
5) High-throughput 시퀀싱을 진행한다.
ChIP 실험에서 회수되는 DNA는 단일 단백질 혹은 단일 복합체에 의해 결합된 영역만을 포함하기에 양이 매우 적다. 더욱, 세포 수가 제한된 상태에서 얻은 ChIP DNA는 더 DNA 양이 적을 수 있어 분석에 매우 어려움이 있었다. 이러한 경우는 50 pg 수준의 샘플 양에도 적용 가능한 ThruPLEX 기술을 적용하면 분석을 용이하게 진행할 수 있다.
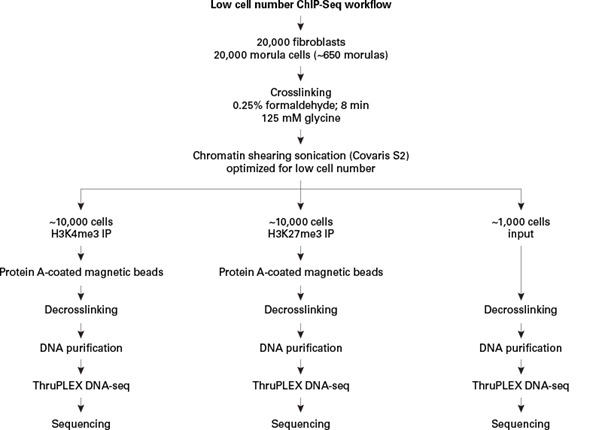 그림 1. 적은 수의 세포를 이용한 ChIP-seq의 실험 과정
그림 1. 적은 수의 세포를 이용한 ChIP-seq의 실험 과정
This is a general workflow for low cell number ChIP-seq. Approximately 20,000 fibroblasts and 20,000 morula cells were each subjected to formaldehyde crosslinking, quenching by glycine addition, and chromatin shearing by sonication. Following shearing, chromatin from fibroblasts or morula cells was divided into subpopulations for both immunoprecipitations, or 10% input controls, followed by decrosslinking, DNA purification, library preparation with ThruPLEX DNA-Seq, and sequencing.
◆ Results
특히 적은 세포로부터 얻은 ChIP DNA의 양은 매우 적기 때문에, 대량 분석을 위한 library를 제작하는 것은 매우 어렵다. 이러한 어려움에도 불구하고, ChIP-seq은 특정 부위에서 작용하는 단백질과 DNA의 상호 작용과 전사 조절을 확인하기 위해 가장 많이 사용되는 기법이 되었다 (Park 2009). 아래에서는 ChIP-seq을 위해 morula embryo 유래의 bovine fibroblast cell과 blastomere 세포 최대 10,000개로부터 H3K4me3 혹은 K3K27me3 항체를 이용해 ChIP DNA를 얻고,
ThruPLEX DNA-Seq Kit를 이용해 라이브러리를 제작하였다. 이렇게 제작된 ThruPLEX 라이브러리는 Illumina
® TruSeq
®를 통해 제작된 라이브러리와 매우 유사하게 나타났다. 10,000개 세포로부터 ThruPLEX
®으로 제작한 ChIP-seq 라이브러리의 reads 수는 107개의 세포로부터 Illumina
® TruSeq
®를 통해 제작하여 얻은 것과 유사하였다 (
그림 2). ChIP-seq의 초기 샘플로 사용된 DNA 샘플은 적은 양의 세포로부터 얻었기에, clonal reads 수도 낮게 생성되었다 (
그림 3).
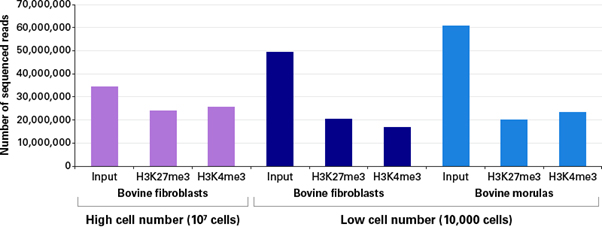 그림 2. 각 조건 별 sequenced reads 수
그림 2. 각 조건 별 sequenced reads 수
Low cell number ChIP-seq samples and inputs were sequenced to similar depths as high cell number ChIP-seq samples
(Panel A), and low cell number ChIP-seq libraries had low percentages of PCR duplicates
(Panel B). Each ChIP and sequencing experiment was performed as a single technical replicate.
ChIP-seq을 위해 TruSeq
®을 이용하는 경우보다 ThruPLEX
®를 이용하는 경우에 약 1,000배 적은 세포를 샘플로 사용할 수 있으며, 총 reads 중 95 - 96%가 bovine genome에 alignment 되어 게놈 전체에서 이상적인 peak를 보였다 (
그림 3).
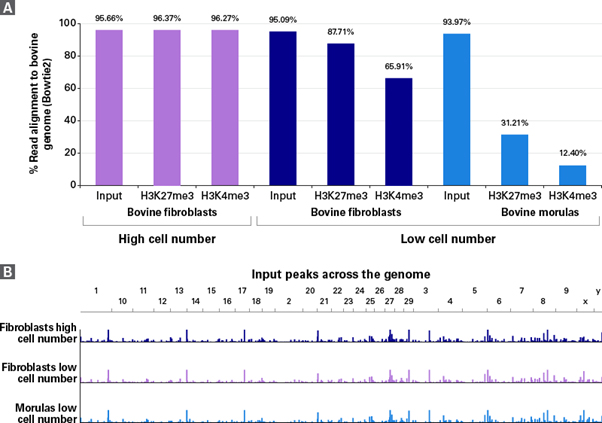 그림 3. Bovine genome에 reads를 alignment한 결과
(Panel A)
그림 3. Bovine genome에 reads를 alignment한 결과
(Panel A) Percentages of reads aligning to the bovine genome, as calculated using the Bowtie2 package, are shown for high cell number input and ChIP samples, as well as low cell number input and ChIP samples.
(Panel B) The peaks identified genome-wide from input DNA are nearly identical when compared between high cell number library preparation using TruSeq and low cell number library preparation using ThruPLEX DNA-Seq
극도로 적은 양의 샘플을 사용한 low cell number ChIP의 경우, 총 reads 수에서 genome에 align한 것이 낮은 비율로 나타났지만 (
그림 3), peak를 통해 확인된 fibroblast의 유전자 수는 매우 비슷하게 보였다 (
그림 4). 배아 발달 단계에서 histone의 methylation 수준이 제한된다는 예상대로, bovine morulas의 ChIP 실험에서는 더 적은 수의 유전자가 검출되었다 (Canovas, Cibelli, and Ross 2012). 이러한 low cell number ChIP-seq 실험 결과는
그림 5에서 보이는 것과 같이 기존의 문헌 및 이전에 진행된 RNA-seq 결과와 아주 높은 연관성을 보였으며, H3K4me3와 결합하는 유전자가 높게 발현되는 반면, H3K27me3와 결합된 유전자는 매우 낮은 transcription level을 보였다 (Martin and Zhang 2005).
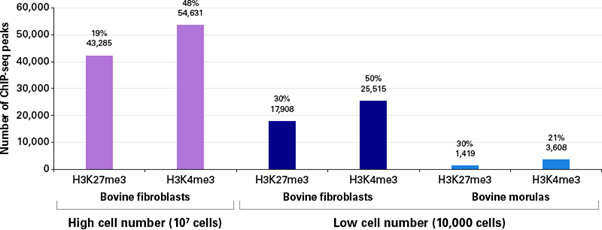 그림 4. Bovine genome에서 보이는 ChIP-seq peaks
그림 4. Bovine genome에서 보이는 ChIP-seq peaks
Above, in black: the number of peaks called to the bovine genome are shown, per ChIP-seq experiment. Above, in red: the percentage of peaks called that specifically overlap genes.
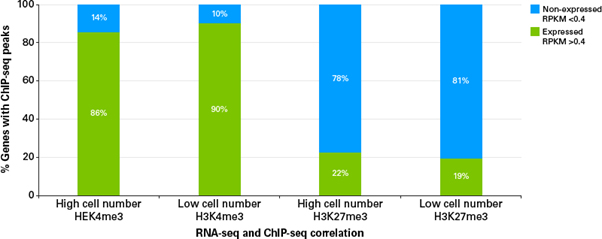 그림 5. ChIP-seq peaks와 transcription level 간의 연관성
그림 5. ChIP-seq peaks와 transcription level 간의 연관성
The percentage of genes with peaks identified in both high and low cell number ChIP-seq and their correlation with transcription level was analyzed. Genes immunoprecipitated with H3K4me3 show high levels of gene expression in prior RNA-seq results. Genes immunoprecipitated with H3K27me3 show reduced transcription.
◆ Conclusion
소량의 ChIP-DNA를 library로 제작하는 것은 굉장히 어렵지만,
ThruPLEX® DNA-Seq Kit를 이용하면 이 어려움을 극복할 수 있다. ThruPLEX 기술은 약 10,000개 이하의 매우 적은 수의 bovine fibroblast와 embryonic cell로부터 얻은 ChIP DNA를 충분히 증폭할 수 있다. 본 실험 데이터에서 확인할 수 있듯이, H3K4me3와 H3K27me3와 결합하는 각각의 DNA로부터 ChIP-seq을 진행했을 때, low cell number ChIP에서도 high cell number ChIP 결과와 매우 높은 연관성을 확인했으며, duplicate reads 또한 낮았다.
ThruPLEX® DNA-Seq Kit는 single tube 내 진행되는 매우 빠르고 간단한 3-step 프로토콜을 자랑하며, 이는 ChIP DNA를 증폭하는 데 있어 기술적 한계를 극복하고 ChIP-seq library 제작 과정의 한 방법으로써 중요하게 활용될 수 있다.
[ References ]
- Barski, A.
et al. High-resolution profiling of histone methylations in the human genome.
Cell 129, 823-37 (2007).
- Canovas, S., Cibelli, J. B. & Ross, P. J. Jumonji domain-containing protein 3 regulates histone 3 lysine 27 methylation during bovine preimplantation development.
Proc. Natl. Acad. Sci. 109, 2400-2405 (2012).
- Cao, Z., Chen, C., He, B., Tan, K. & Lu, C. A microfluidic device for epigenomic profiling using 100 cells.
Nat. Methods 12, 959-962 (2015).
- Dahl, J. A. & Collas, P. μChIP-a rapid micro chromatin immunoprecipitation assay for small cell samples and biopsies.
Nucleic Acids Res. 36, e15 (2008).
- Dennis, G.
et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery.
Genome Biol. 4, P3 (2003).
- Farnham, P. J. Insights from genomic profiling of transcription factors
. Nat. Rev. Genet. 10, 605-616 (2009).
- Johnson, D. S., Mortazavi, A., Myers, R. M. & Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions.
Science 316, 1497-1502 (2007).
- Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2
. Nat. Methods 9, 357-359 (2012).
- Martin, C. & Zhang, Y. The diverse functions of histone lysine methylation.
Nat. Rev. Mol. Cell Biol. 6, 838-849 (2005).
- Park, P. J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669-680 (2009).
- Robinson, J. T.
et al. Integrative genomics viewer.
Nat. Biotechnol. 29, 24-26 (2011).
- Ross, P. J.
et al. Activation of bovine somatic cell nuclear transfer embryos by PLCZ cRNA injection.
Reproduction 137, 427-437 (2009).
- Salmon-Divon, M., Dvinge, H., Tammoja, K. & Bertone, P. PeakAnalyzer: Genome-wide annotation of chromatin binding and modification loci.
BMC Bioinformatics 11, 415 (2010).
- Zhang, Y.
et al. Model-based Analysis of ChIP-Seq (MACS).
Genome Biol. 9, R137 (2008).
[원문] Low cell number ChIP-seq using the ThruPLEX DNA-Seq Kit as a tool for epigenetic profiling